Amines can participate in E2 reactions to form alkenes. However, just like the OH group of alcohols, the NH2, or any other amino group, is a quite strong base and needs to be first converted into a good leaving group.

This is achieved by methylation of the nitrogen with an excess of methyl iodide. The product of this reaction is a quaternary ammonium salt, which is a good leaving group in the form of a neutral amine:

The E2 elimination leading to an alkene is accomplished with a hydroxide ion, which is formed in situ when the substrate is heated with Ag2O:

This is called the Hofmann elimination reaction, and it should remind you the principles of the Zaitsev and Hofmaan eliminations. We will get to those rules right after first discussing the general idea behind this method of elimination.
So, let’s answer a couple of important questions.
First, why not just use a hydroxide instead of Ag2O and where is the hydroxide actually coming from when Ag2O is added?
The argument for not adding a hydroxide directly can be the fact that iodide, being in the 5th row of the periodic table, is a large ion, and its negative charge blocks the access of the hydroxide ion.

As far as why the iodide itself does not perform the elimination reaction, we need to keep in mind that it is not such a good base. In fact, quite the opposite, the HI is a quite strong base, meaning the iodide is a weak base-good leaving group.
Now, where is the OH coming from, and how come when formed in situ, it is capable of doing the E2? Remember that silver has a high affinity to halide ions, and their precipitates are some of the famous wones e learn in general chemistry. So, the silver oxide captures the iodide, forming a nice precipitate and replacing the counterion of the quaternary ammonium salt with a hydroxide.
After this, the hydroxide ion, being next to the β hydrogen,s can perform the E2 elimination:

The Regiochemistry of Hofmann Elimination
Remember, the E2 elimination favors the more substituted alkene when a non-hindered base is used:

This is known as the Zaitsev’s rule, which is explained by the stability of the more substituted alkene. Now, it is interesting that the regioselectivity of quaternary ammonium salt elimination follows a different path, and the less substituted alkene is the major product.
For example, the elimination of 2-aminopentane produces 1-pentene as the major product:
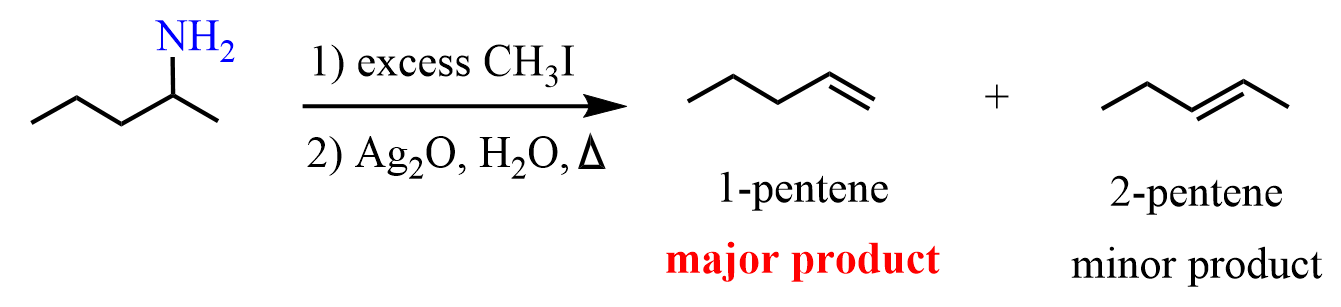
This looks counterintuitive, but the good news is that we can use both the electronic and steric arguments to explain this regiochemistry.
The Electronic Argument
Let’s start with electronics. What we need to know first is that the amino group of the quaternary ammonium salt is not an excellent leaving group. It is, for example, a poorer leaving group than chloride, bromide, and iodide. So, when the OH attacks the β hydrogen, the amino group is not expelled precisely at the same time as the double bond is being formed, which is what we know about the traditional E2 mechanism. Instead, the amino group is still in place when the C-H bond is being broken, and therefore, the lone pair resides on the carbon rather than making a new π bond.
Below is the comparison of both possibilities forming the Zaitsev and Hoffmann products. The reaction on the left is more favorable since the negative charge is concentrated on a less substituted carbon than it would if the Zaitsev product (on the right) were to predominate.

Now, if we compare this process happening on the more substituted- and the less substituted carbons, we can see the preference for placing a partial negative charge on the less substituted carbon. Remember, less substituted carbanions are more stable.
Therefore, the hydrogen is removed from the less substituted β-carbon in order to form the more stable carbanionlike transition state, which leads to the Hofmann product.
The Steric Argument
To understand this explanation, first, recall that E2 elimination requires an antiperiplanar arrangement of the leaving group and β hydrogen:

Now, let’s compare the energies of the conformations when either the β hydrogens of the more or the less substituted carbons are aligned at 180o with the ammonium group.
The geometry suitable for the elimination involving the more substituted β position is associated with a gauche interaction when the β hydrogen on the more substituted carbon is placed opposite to the leaving group.
On the other hand, any of the protons of the methyl group allows to avoid the unfavorable gauche interaction and lowers the transition state for the formation of the less substituted alkene:

In the end, it is worth mentioning that this regiochemical outcome is also observed in the elimination of alkyl fluorides. The fluoride is a poor leaving group and favors the Hofmann elimination product regardless of whether a hindered or unhindered base is used:
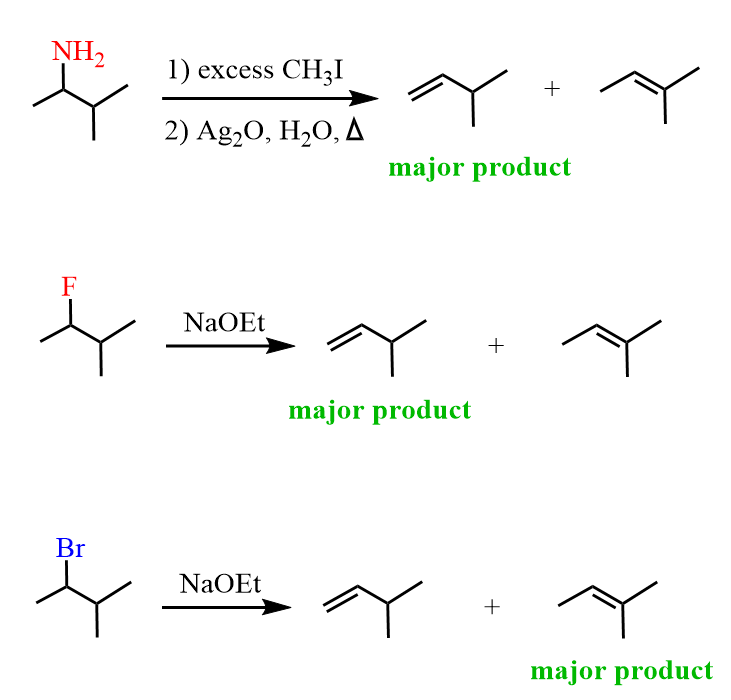
There are a couple of explanations for this regioselectivity of alkyl fluoride eliminations. The C-F bond is stronger than other C-halogen bonds, and the fluoride is a poor leaving group. Because of this, the elimination does not follow the classical E2 pattern, where the abstraction of the ß-hydrogen and loss of the leaving group happen simultaneously. Instead, the proton is removed, leaving behind a carbanion which then expels the F– by forming a π bond.
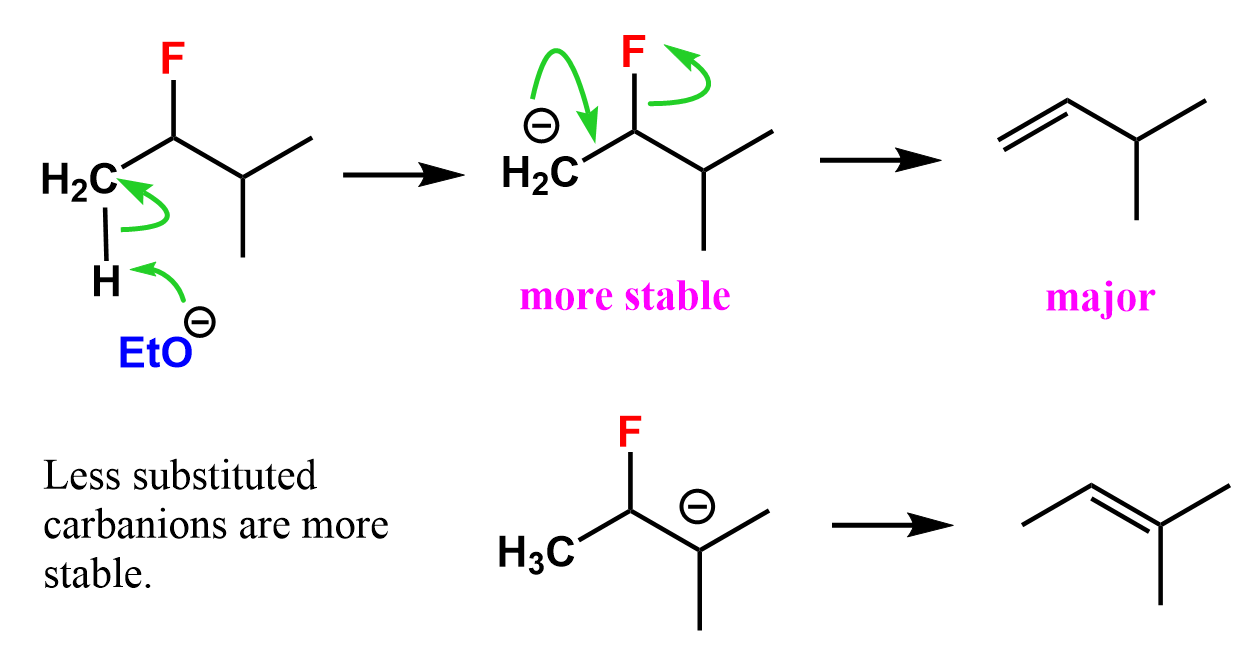
This is known as the E1cB mechanism, which is mainly covered in Organic Chemistry II, so there’s no need to worry about the finer details just yet. The key question is: Why is the less substituted alkene the major product? The answer lies in the greater stability of the less substituted carbanion.
Unlike carbocations, carbanions become less stable as more alkyl groups are attached to the negatively charged carbon. This is because alkyl groups are electron-donating, which increases electron density on an already negatively charged center, making it less stable.
Whether the reaction follows a purely E2 or E1cB mechanism, the β-carbon develops significant negative character in the transition state due to the poor leaving ability of the C–F bond. This favors deprotonation at the less substituted β-carbon, leading to the formation of the less substituted alkene (Hofmann product).
In reality, the mechanism likely lies somewhere between the E2 and E1cB pathways.
Check Also
- Preparation of Amines
- The Gabriel Synthesis of Primary Amines
- The Hofmann Elimination of Amines and Alkyl Fluorides
- Imines from Aldehydes and Ketones with Primary Amines
- Enamines from Aldehydes and Ketones with Secondary Amines
- The Reaction of Amines with Nitrous Acid
- Reactions of Amines Practice Problems