There are a few main strategies for preparing epoxides, and the good news is that we have seen all of them in earlier chapters. Therefore, this post is a review of reactions leading to epoxidation with additional emphasis on the stereochemistry of these reactions, supported by some practice problems.
Oxidation of Alkenes with Peroxycarboxylic Acids
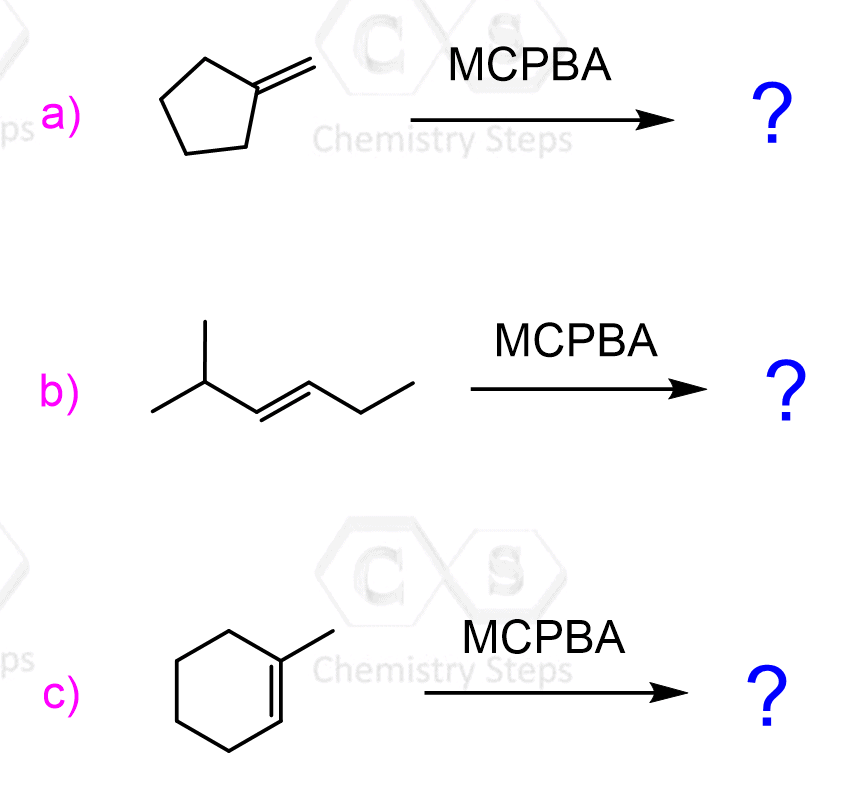
One of the most common ways of epoxidation is the addition of an oxygen atom to an alkene with peroxycarboxylic acids (sometimes referred to as peroxyacids):

Representative examples of peroxycarboxylic acids are MCPBA (meta-chloroperoxy benzoic acid) or Peroxyacetic acid, which we have seen in the anti-dihadroxylation of alkenes.
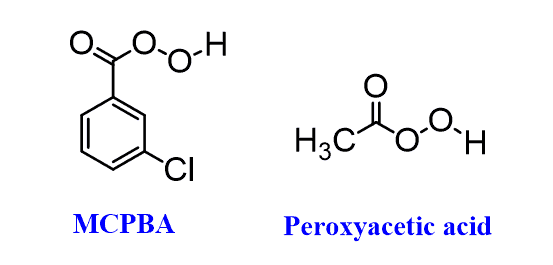
The reason for the popularity of mCPBA is that, unlike many peroxycarboxylic acids, which need to be prepared right before the reaction by mixing a carboxylic acid and hydrogen peroxide, it is a quite stable crystalline solid and can be purchased commercially.
This does not, however, exclude it from the characteristic of the danger of detonation if not handled properly.
Now, the fact that peroxides have low stability indicates that they are very reactive, and this is explained by the presence of the weak O-O bond. This, in the presence of a weak π bond, is a good combination to drive the epoxidation reaction forward.
The mechanism of epoxidation by Peroxycarboxylic Acids
The oxygen in the peroxide is electron-deficient and is attacked by the p electrons of the π bond. At the same time, the lone pair of the oxygen also acts as a nucleophile, attacking one of the carbon atoms of the alkene.

The weakness of the O-O bond makes the bond-cleavage easier, thus acting as a driving force for the epoxidation.
This mechanism is similar to the formation of halonium ion and oxymercuration, which we have also discussed in the other addition reactions of alkenes.
An alternative, short version of the reaction can be drawn when the nucleophilic attack of the peroxide originates from the O-H bond instead of the lone pair of the oxygen.

In any case, one thing is for sure – epoxidation is a concerted mechanism, meaning that all the bonds are formed and broken simultaneously (recall the old and sweet SN2 mechanism).
There are two arguments we can bring to support the concerted nature of the mechanism.
First, carbocation rearrangements are not observed, and second, the stereochemistry of the reaction is a stereospecific syn-addition. Let’s look at this in a little more detail.
The Stereochemistry of Epoxidation by Peroxycarboxylic Acids
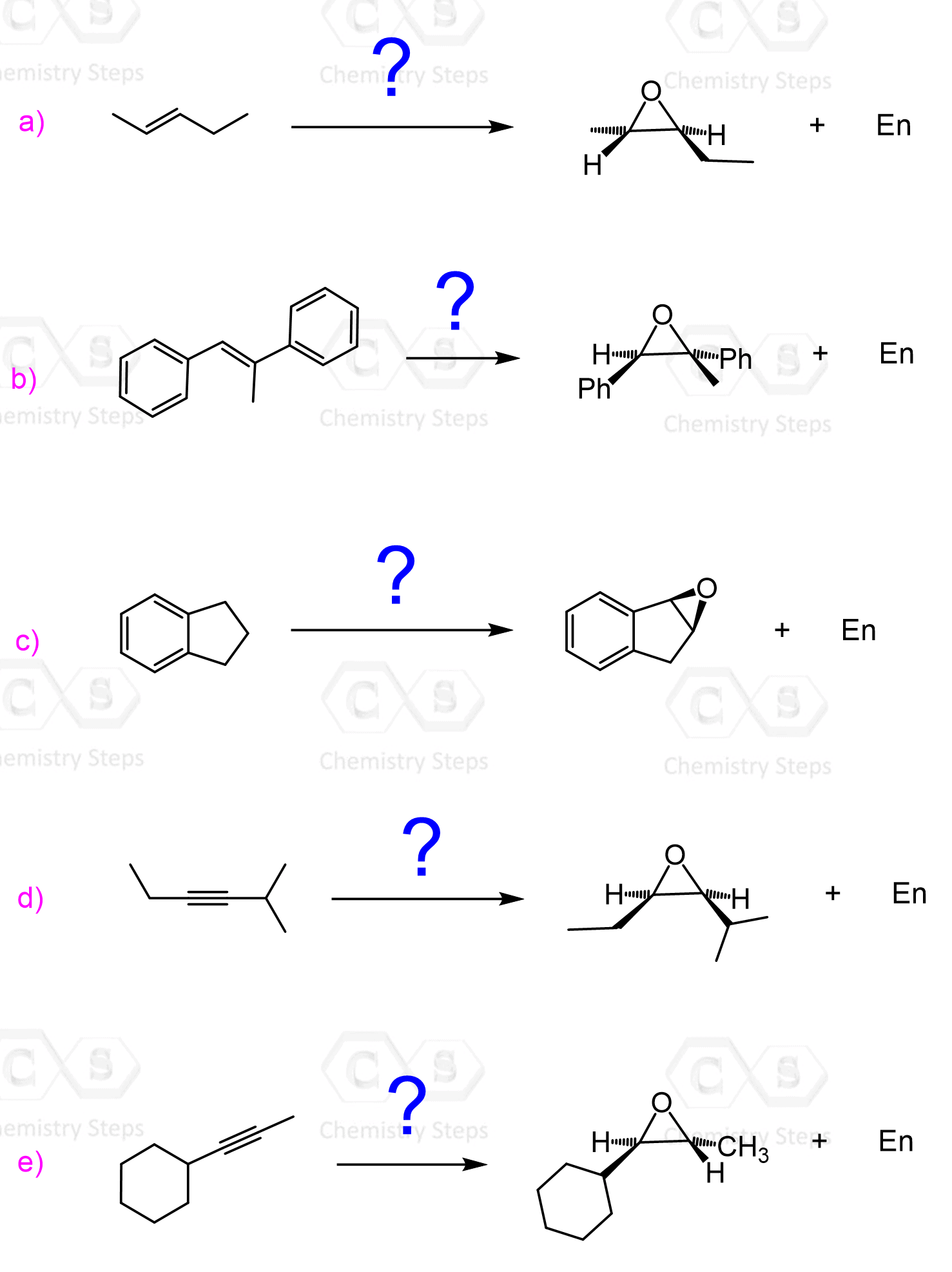
The epoxidation is a syn addition, and the stereochemistry of the product is determined based on the structure of the alkene. Cis alkenes give cis epoxides, and trans alkenes give trans epoxides.
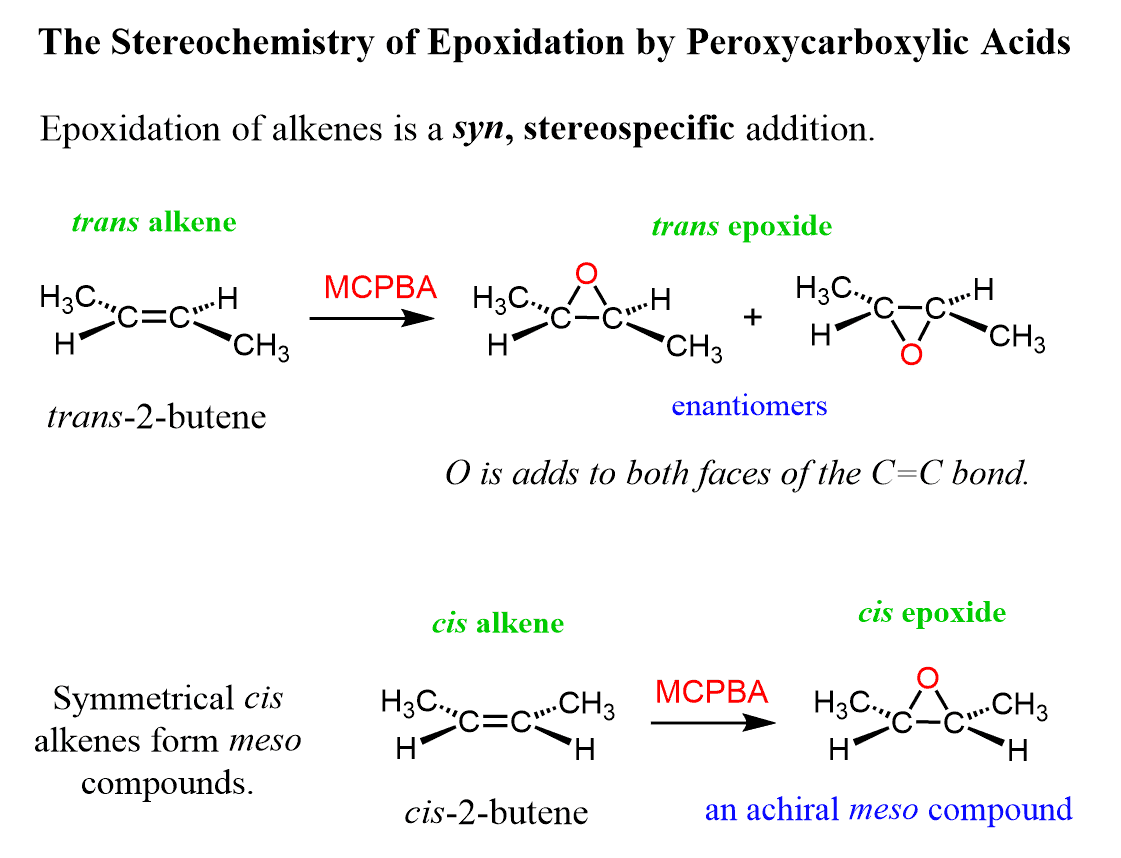
Remember, when the stereochemistry of the product is dictated by the stereochemistry of the substrate, the reaction is said to be stereospecific, i.e. it has no choice-cis gives cis, trans gives trans regardless of their stability.
The epoxide can react with the alkene from both faces; therefore, the oxygen can add to either face of the alkene.
As a result, a mixture of enantiomers is obtained when substituted alkenes react with acid peroxides:

You can selectively produce one of the enantiomers in excess by enantioselective epoxidation, for example, by Sharpless epoxidation, but we will skip this for now, as it is often not needed in an undergraduate course of Organic Chemistry.
As usual, watch for meso compounds, as not every compound with stereogenic centers is going to be chiral. For example, when a symmetrical cis alkene is used, the corresponding meso epoxide is obtained:
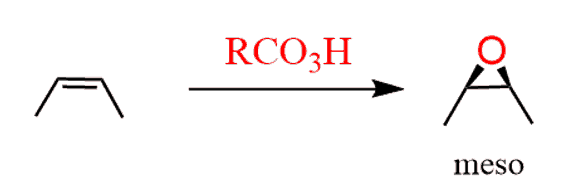
Notice that this is the same reaction/product shown a little earlier above.
Epoxides by Cyclization of Halohydrins
Another approach for preparing epoxides is the intramolecular SN2 reaction of halohydrins upon treatment with a strong, non-nucleophilic base such as sodium hydride, NaH.

This is a variation of the Williamson ether synthesis, and what happens is that the sodium hydride deprotonates the alcohol, converting it into a better nucleophile.

This is an intramolecular SN2 reaction, and the OH and Br must be in a trans configuration in order to accomplish the proper orbital alignment. We can see this better by drawing the chair conformations of the molecule.
Not only must the groups be trans, but they also need to be in an axial position because there is no good alignment of HOMO and LUMO orbitals for the substitution to occur:

Although the equilibrium is favored for the conformation with the two groups being equatorial, the diaxial conformation quickly undergoes a substitution reaction, thus moving the process forward.
Check also this article on the SN2 and E2 reactions of cycloalkanes, where we discuss the importance of the leaving group being in the axial position.
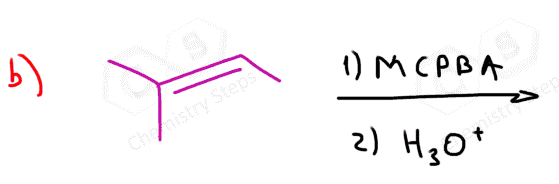
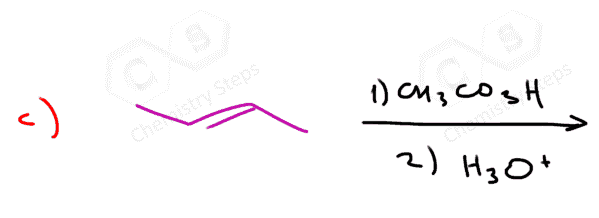
You have your trans and cis alkenes misnamed I believe.
Thanks for spotting that! Fixed.
Very helpfull, simple and explanatory. THANKS