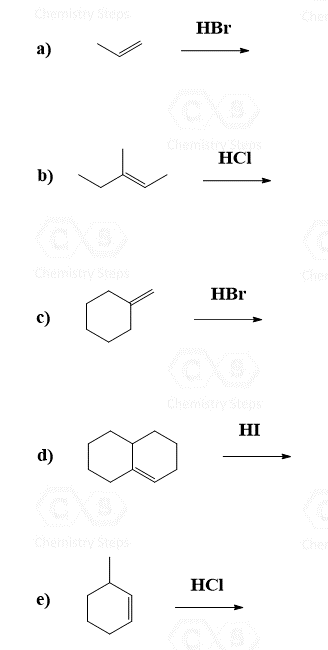
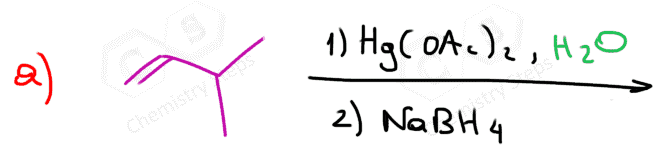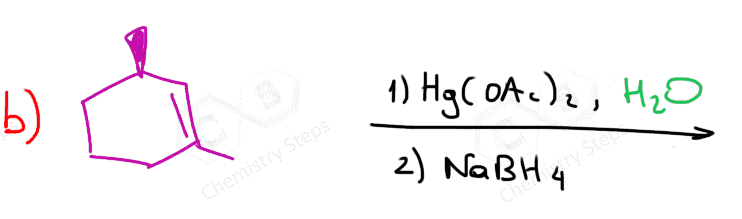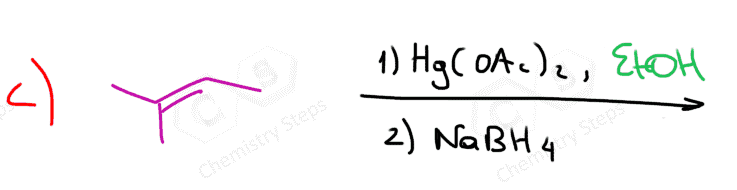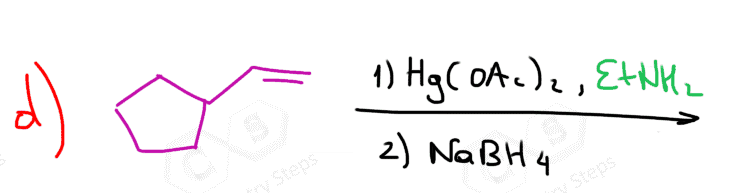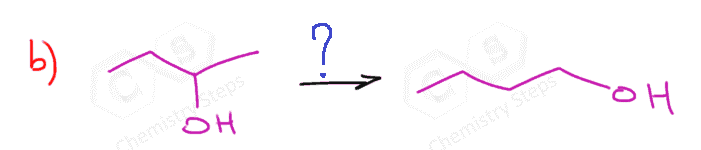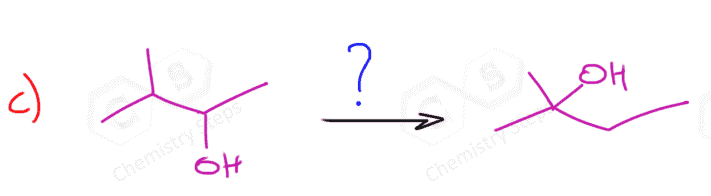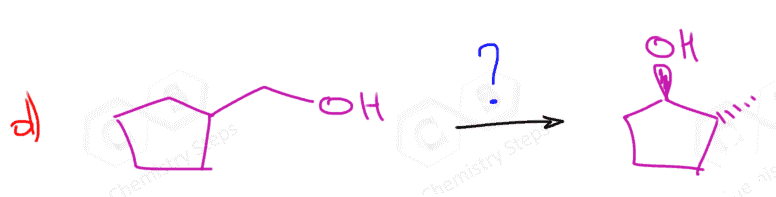Addition of HBr, HCl, and HI to Alkenes
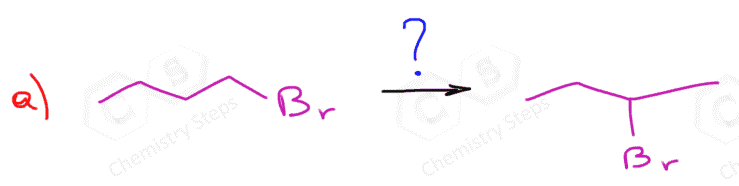
The regiochemistry of regioselectivity of a reaction is the preferential formation of one out of two or more possible regioisomers. For example, when an unsymmetrical akene is reacted with a hydrohalic acid such as HBr, HCl, and HI, the halogen can theoretically end up either on the more substituted carbon or the less substituted carbon of the double bond:
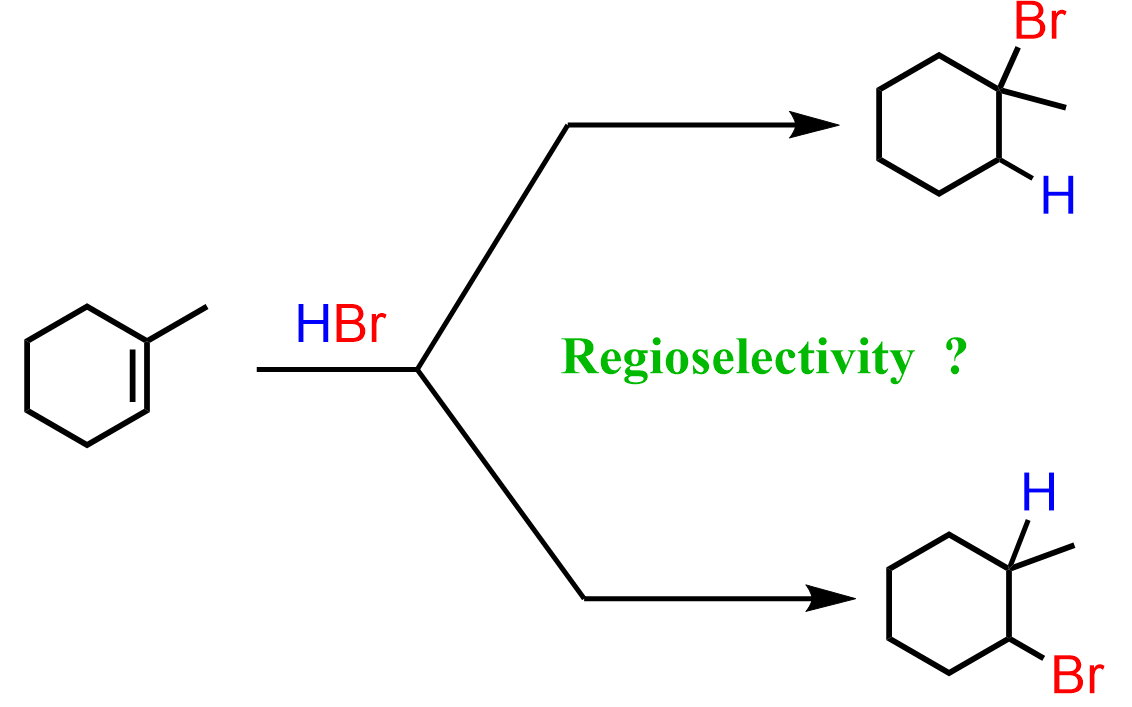
If in a given reaction, one of the pathways is predominant, then we say that the reaction is regioselective. In hydrohalogenation reactions, the halogen most often adds to the more substituted carbon of the double bond (Markovnikov’s rule) unless HBr is used in the presence of a peroxide which changes the reaction to a radical mechanism and the Br adds to the less substituted carbon:
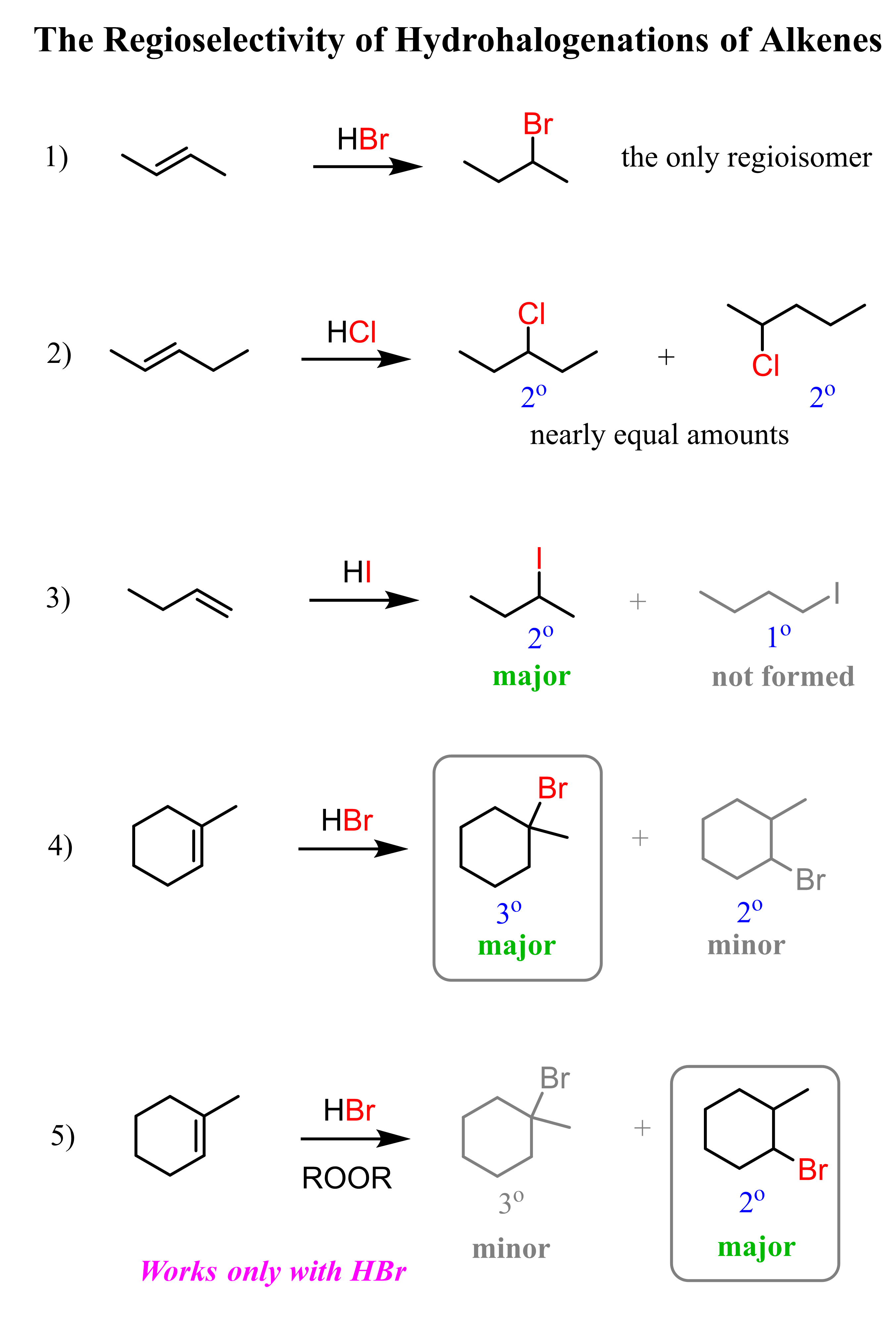
The reason for this is that in the radical mechanism, Br radical adds to the double bond first, forming the more substituted radical intermediate, whereas in the ionic mechanism, the protonation occurs first, favoring the stability of the more substituted carbocation.
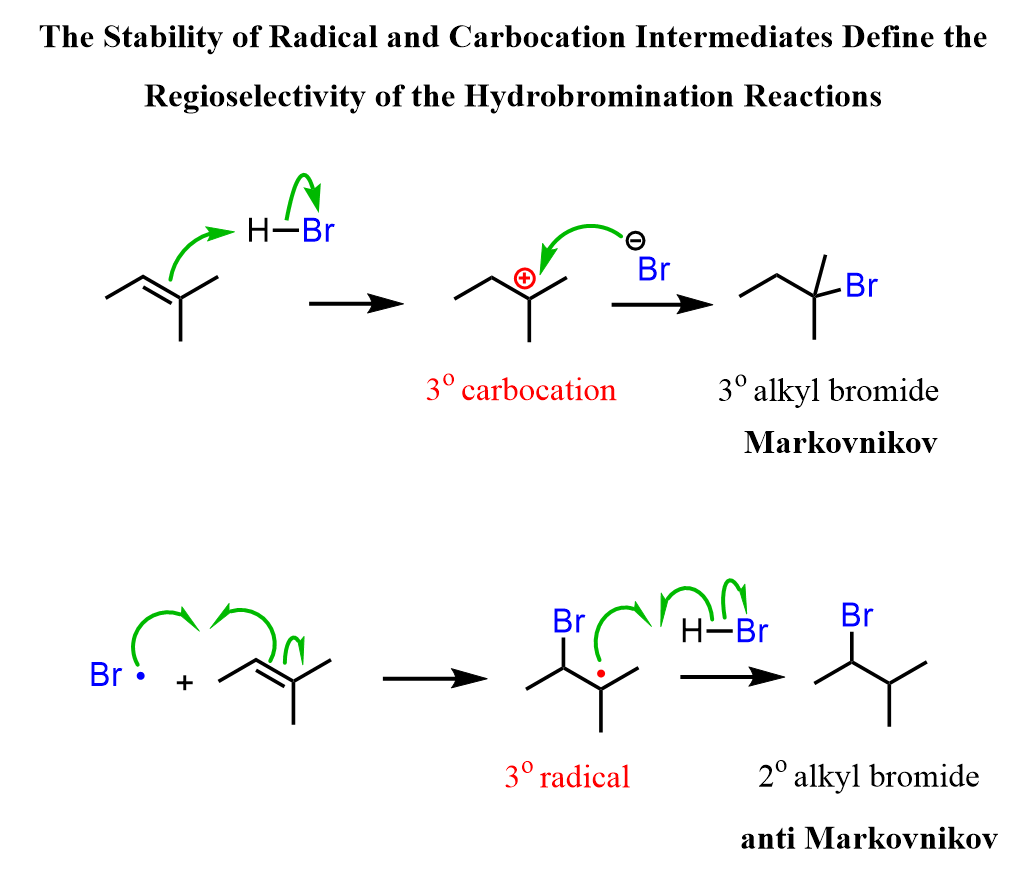
Recall that carbocations and radicals have the same pattern of stability – the more substituted, the more stable.

Keep in mind that this anti-Markovnikov hydrohalogenation is only possible with HBr and not with HCl or HI.
We won’t go too much into the details of the mechanisms in this post, as the focus here is to summarize the patterns of regioselectivity in electrophilic additions to alkenes. For detailed discussions, you can click on the corresponding articles for each reaction.
Rearrangements in Hydrohalogenation of Alkenes
Because of the carbocation intermediate, rearrangements can occur during hydrohalogenation reactions. Always check if the carbocation has a way of transforming into a more stable one via hydride or methyl shifts or ring expansion rearrangement. For example, the major product of the following hydrohalogenation is the tertiary alkyl halide instead of the expected secondary product:
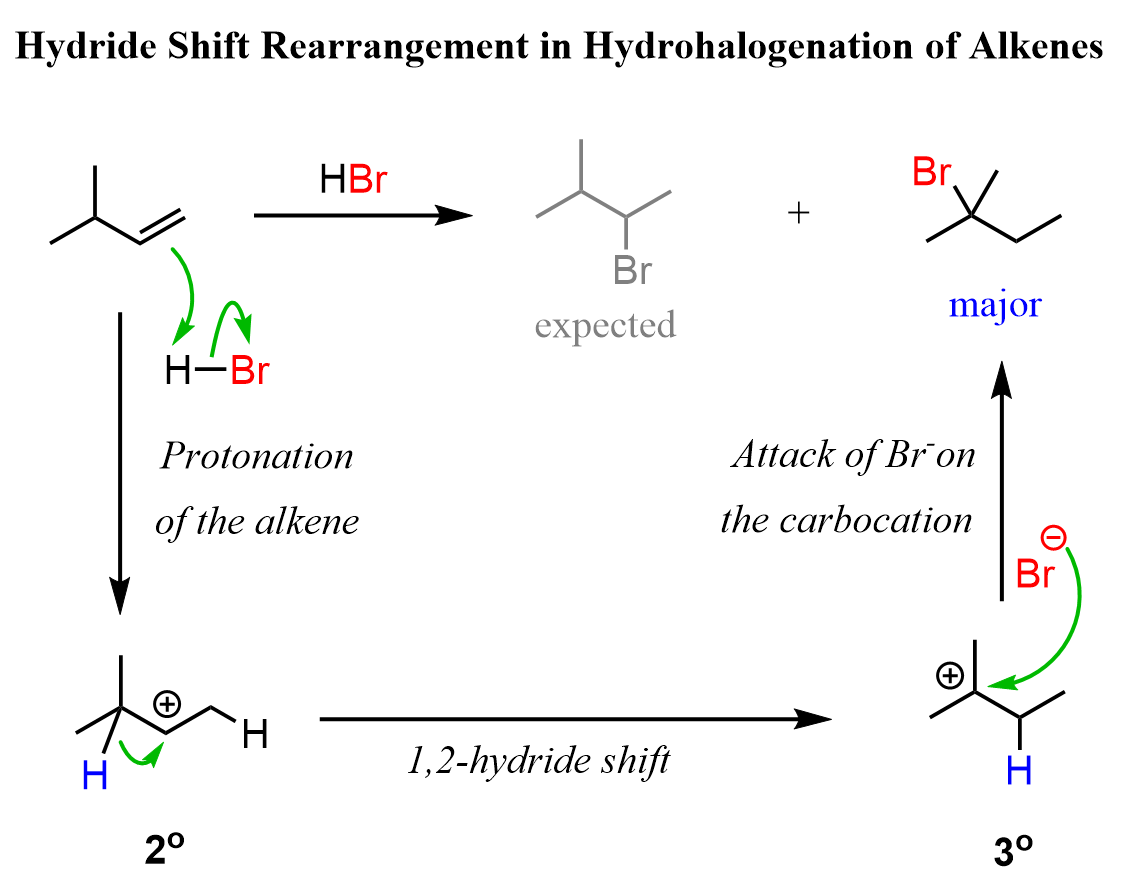
To summarize, the hydrohalogenation of alkenes, we can say that:
1) Regioselectivity is irrelevant for symmetrical alkenes
2) For carbocations, not prone to rearrangements, hydrohalogenation follows Markovnikov’s rule
3) Radical addition of HBr to alkenes gives the anti-Markovnikov product (no rearrangement)
4) When possible, rearrangements do occur
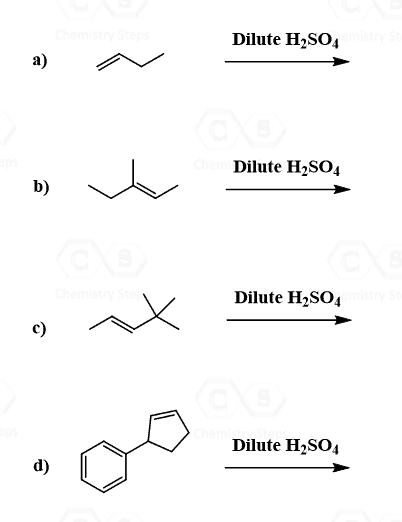
Addition of Water to Alkenes
From the regiochemical perspective, the addition of water to alkenes is seen as adding an OH group to the double bond. There are three main ways of doing it:
1) Acid-Catalyzed Hydration, 2) Oxymercuration-Demercuaration, and 3) Hydroboration-oxidation
The first two are used for a Markovnikov hydration of the alkene, meaning they add the OH on the more substituted carbon of the double bond. In contrast, hydroboration oxidation is used for the anti-Markovnikov hydration of the alkene. The oxymercuration-demercuration is used as an alternative to prevent rearrangements in acid-catalyzed hydration:
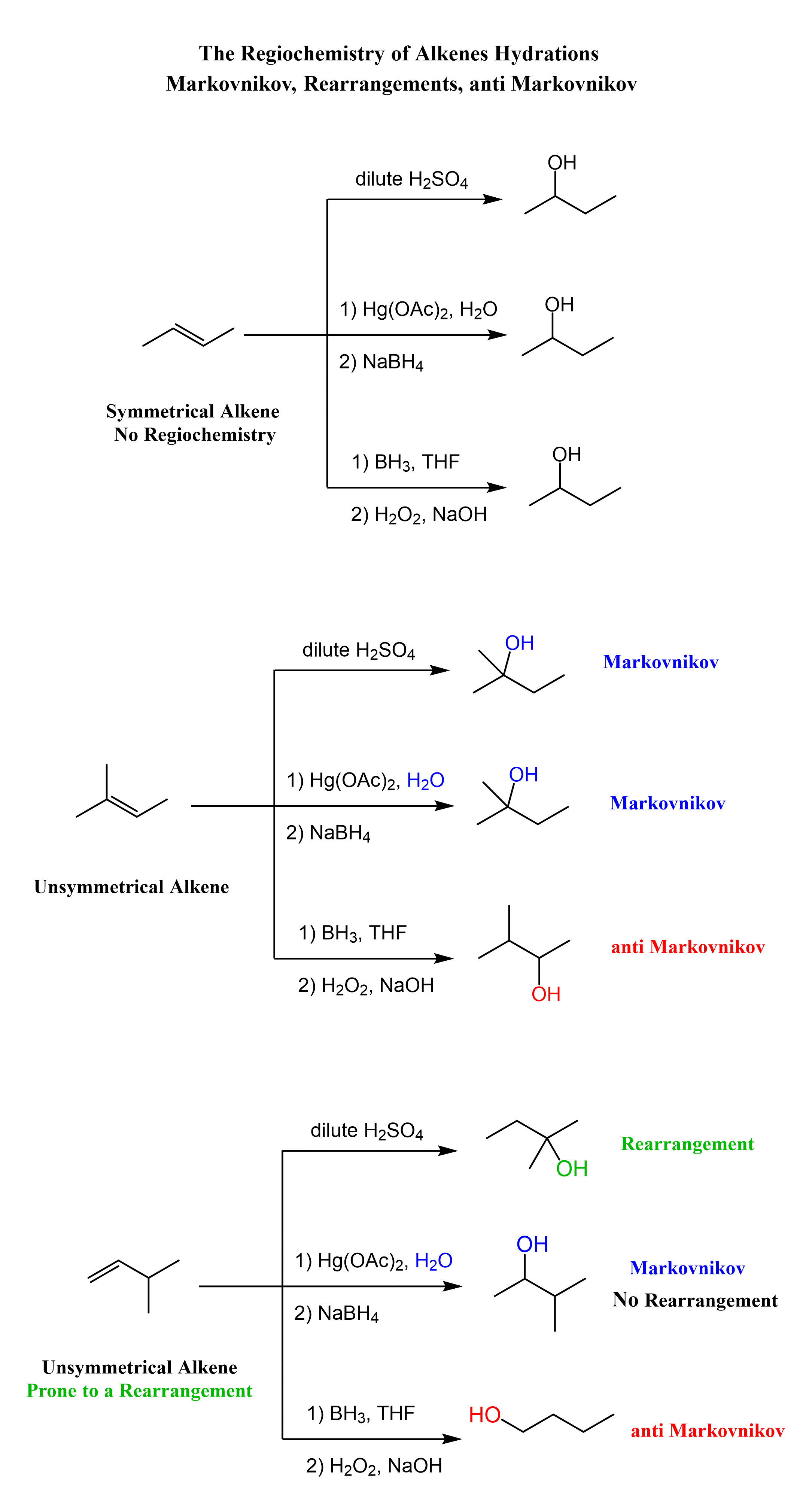
The key difference, compared to acid-catalyzed hydration, is that the intermediate in the oxymercuration-demercuration reaction is the mercurinium ion which does not undergo a rearrangement. What is also important is that the water attacks the more substituted carbon of the mercurinium ion, thus the corresponding alcohol is formed (Markovnikov addition):
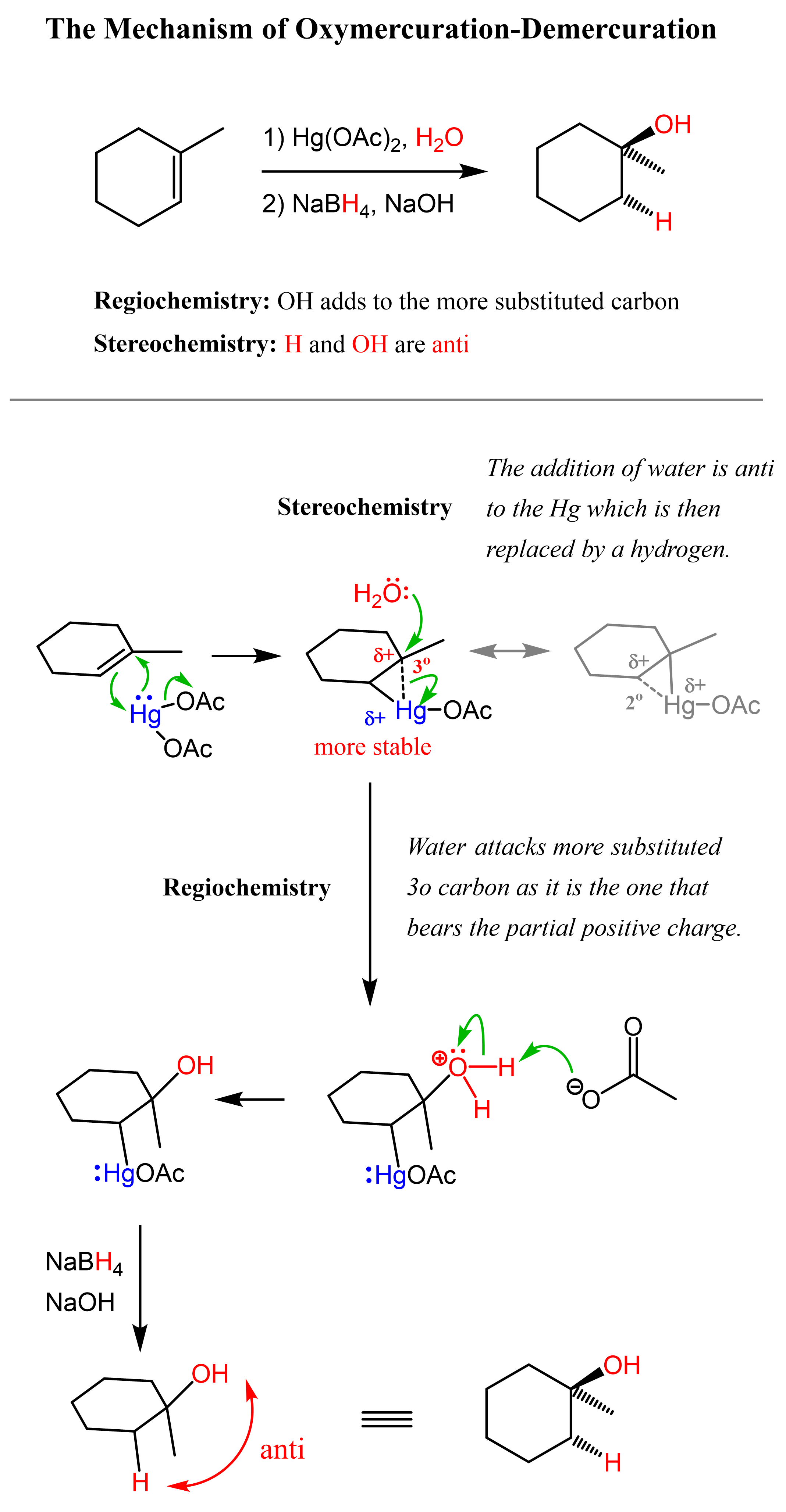
In the last part, the mercury is then removed using sodium borohydride (NaBH4), which reduces the acetoxymercury group and replaces it with its own hydrogen. This is described as demercuration, and it is believed to follow a radical mechanism that is not covered in undergraduate courses.
You can find all the details, including the mechanism and stereochemistry of these reactions, in the corresponding articles linked in this discussion.
Addition of Alcohols to Alkenes
Alcohols can be added to alkenes much like the addition of water, using acid or Hg(OAc)2 catalysts. The adding component in this reaction is the OR group, and therefore, the product is an ether.

When using the mercury catalyst, the reaction is called alkoxymercuration, and the catalyst is again added to prevent any possible. For example, when 3-methylbut-1-ene is reacted with methanol under acidic conditions, 2-methoxy-2-methylbutane is obtained instead of 2-methoxy-3-methylbutane, which would be the Markovnikov product:
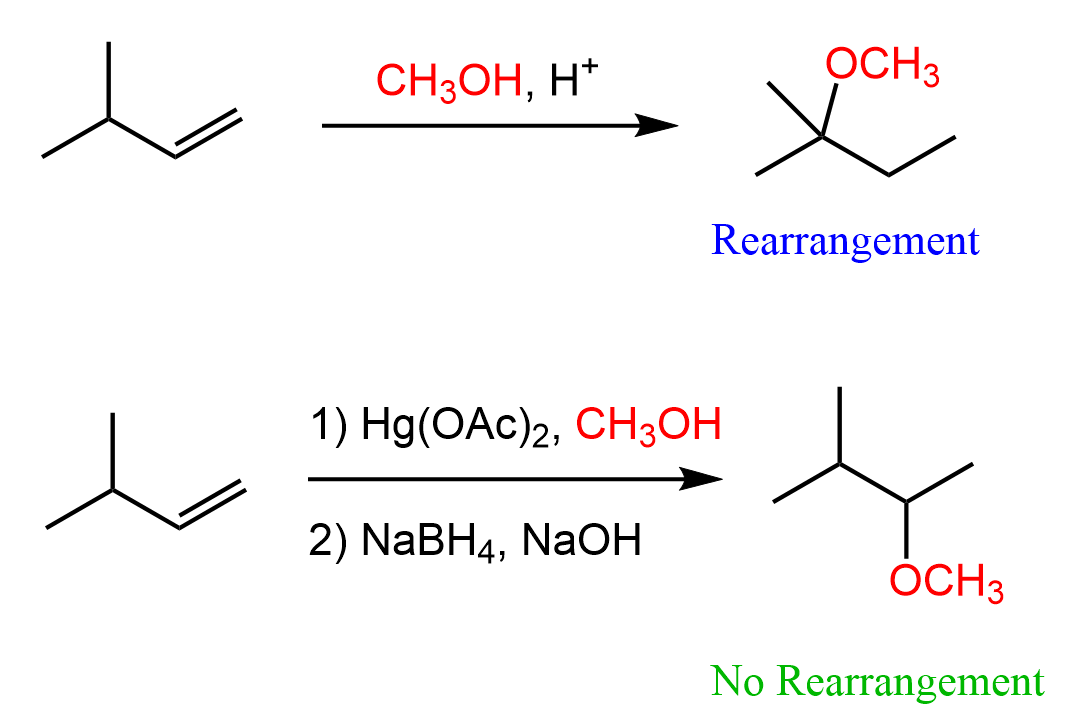
The mechanism is similar to the one in oxymercuration-demercuration, and you can find it in this article.
Halogenation of Alkenes
Alkenes readily react with halogens because of the electron-rich feature of the double bond. In fact, the reaction with bromine is a standard test for the presence of a π bond. Bromine is a dark red liquid, and upon reacting with a double bond, turns colorless.
If water or other nucleophiles such as amines, alcohols or thiols are not added, the product is a vicinal dihalide – a compound bearing the halogens on adjacent carbons (vicinus, Latin: adjacent). These are also called 1,2-dihalides:

The halogenation is an exclusive anti-addition to the double bond. For example, the addition of bromine to cyclohexene produces trans-1,2-dibromocyclohexane, and cis-1,2-dibromocyclopentane is not observed:
![]()
This is explained by the halonium ion intermediate, which is a three-membered ring that undergoes a nucleophilic attack by the halide ion in an SN2 fashion:

Notice that the product is a racemic mixture of enantiomers because the initial addition of the Br to the alkene occurs from both faces of the double bond, forming two enantiomers of the bromonium ion.
Watch out for meso compounds. Not every dihalide with chiral centers is going to be chiral:

Check this article for more details about the stereochemistry of alkenes halogenation reactions, and for now, let’s address the regiochemistry. Unless other nucleophiles such as alcohols, amines, and thiols, capable of attacking the intermediate halonium ion, are added, the product of the reaction is always a 1,2-dihalide. In the presence of the mentioned nucleophiles, the regiochemistry becomes relevant because the halogen and the nucleophile can theoretically add to any of the carbon atoms.

However, it has been shown that the nucleophile adds to the more substituted carbon, and the halogen ends up being on the less substituted carbon:

This is explained by the stability of the energy differences between the two possible transition states:
![]()
The first transition state has a partial positive charge on a more substituted carbon, making it more stable. This is similar to what we observe in the oxymercuration reaction, where the stability of one of the “transition states” dictates the addition of water or other nucleophiles to the more substituted carbon of the mercurinium ion:

When other nucleophiles such as alcohols, amines, thiols etc. are added to the reaction mixture, they react with the halonium ion in the same way:
Dihydroxylation of Alkenes
Dihydroxylation is the addition of two OH groups to the double bond, and the product of these reactions is a 1,2-diol.

So, as far as the regiochemistry, there is not much to discuss. However, the addition of the OH groups can be syn or anti, depending on the reagent in use. The anti-dihydroxylation of alkenes is achieved by converting them not epoxides, followed by acid or base-catalyzed ring-opening of the ring:
![]()
To convert alkenes into cis-diols by syn dihydroxylation, they are reacted with a basic solution of potassium permanganate (KMnO4) or Osmium tetroxide (OsO4):

Check the linked article for details about the mechanism and stereochemistry of these reactions.