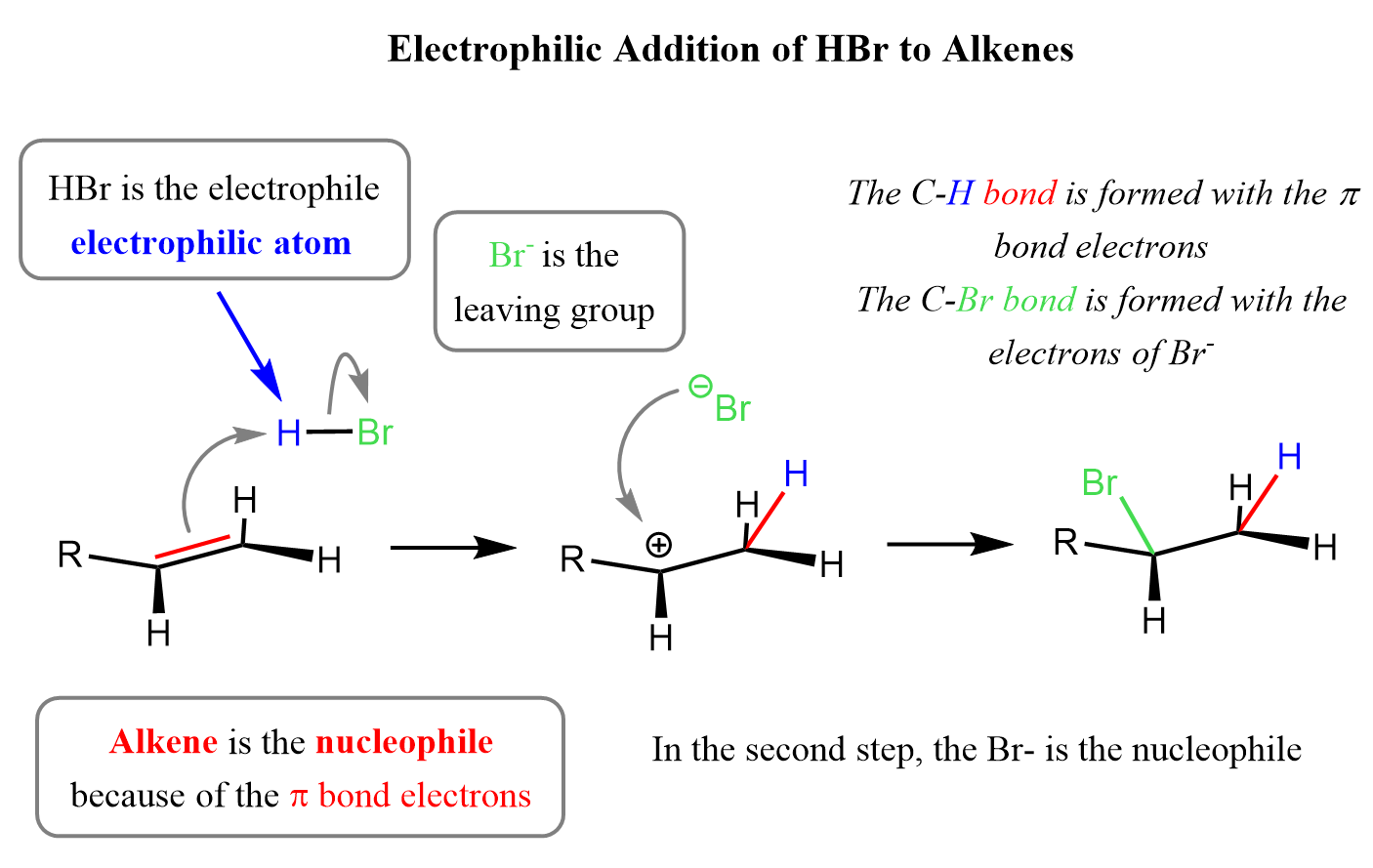
In the introductory post about electrophilic addition reactions to alkenes, we emphasized that they are electron-rich species because of the two extra electrons brought in by the π bond. This feature makes them the nucleophile, or Lewis base in a broader definition, in most of the addition reactions, such as those with hydrogen halides (HCl, HBr, HI). It is the other reactant that is the electrophile, hence the name electrophilic addition:
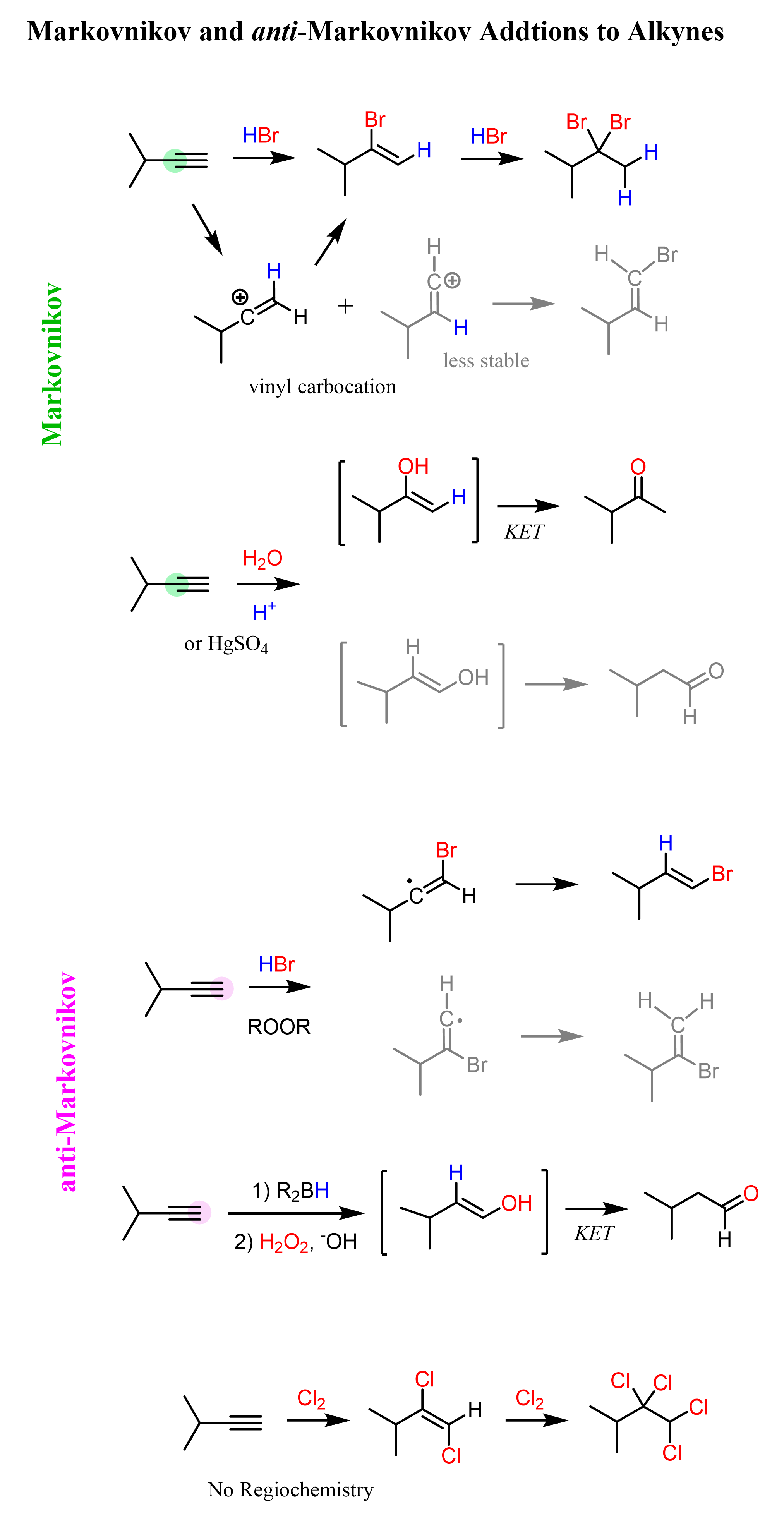
Now, alkynes have an extra π bond compared to alkenes, and there is no apparent reason not to expect them to undergo the same types of electrophilic addition reactions. Just like alkenes, they react with HX acids, halogens, and water; they can be hydrated and subjected to ozonolysis. They do, and when enough reactant is added, some of these additions occur twice because of the second double bond in the molecule:
The Mechanism and Rate of Alkyne Reactions
The mechanism of electrophilic additions to alkynes follows the same pattern as we saw in the reactions of alkenes: formation of a carbocation followed by a nucleophilic addition.
One may expect the reaction of alkyne to go faster as they have two π bonds and therefore, a greater electron density. While this argument is not wrong in and of itself, we also need to compare the stability of the intermediate carbocations. In the case of alkenes, we have a “regular” carbocation with an sp2-hyubridized carbon atom which is stabilized by the bonded alkyl groups. The protonation of a triple bond, on the other hand, forms a vinyl carbocation, which is believed to be sp-hybridized and much less stable than alkyl carbocations:
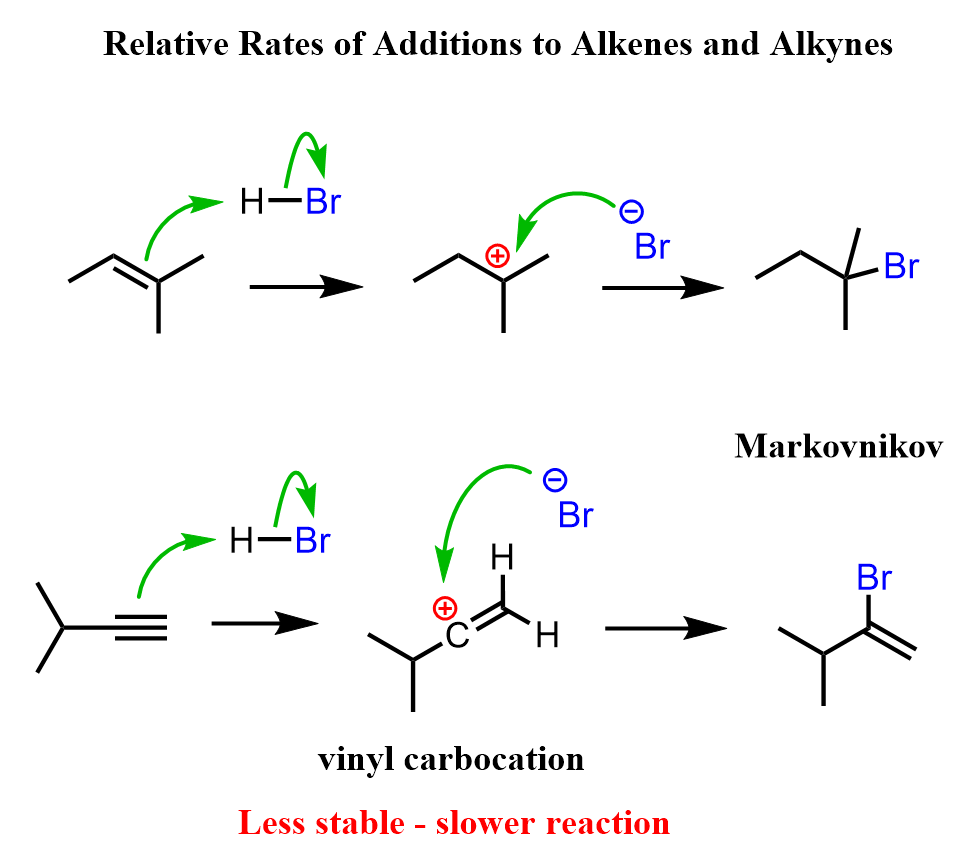
The instability of the vinyl carbocation slows down the reactions to alkynes, and it is actually a subject of an argument whether the electrophilic additions to alkynes occur in this traditional mechanism with carbocation intermediates. With that said, it is still a widely accepted mechanism, especially in undergraduate organic chemistry classes, and this is what we will be using throughout this chapter on the reactions of alkynes. In general, electrophilic additions to alkynes are slower than those of alkenes.
The Regiochemistry of Electrophilic Additions to Alkynes
Like in the reactions of alkenes, we still classify the additions to alkynes as Markovnikov or ani-Markovnikov. Most reactions, such as hydrohalogenation and hydration of alkynes, result in a Markovnikov addition because, like other carbocations, vinyl carbocations are stabilized by the connected alkyl groups: the more substituted, the more stable.
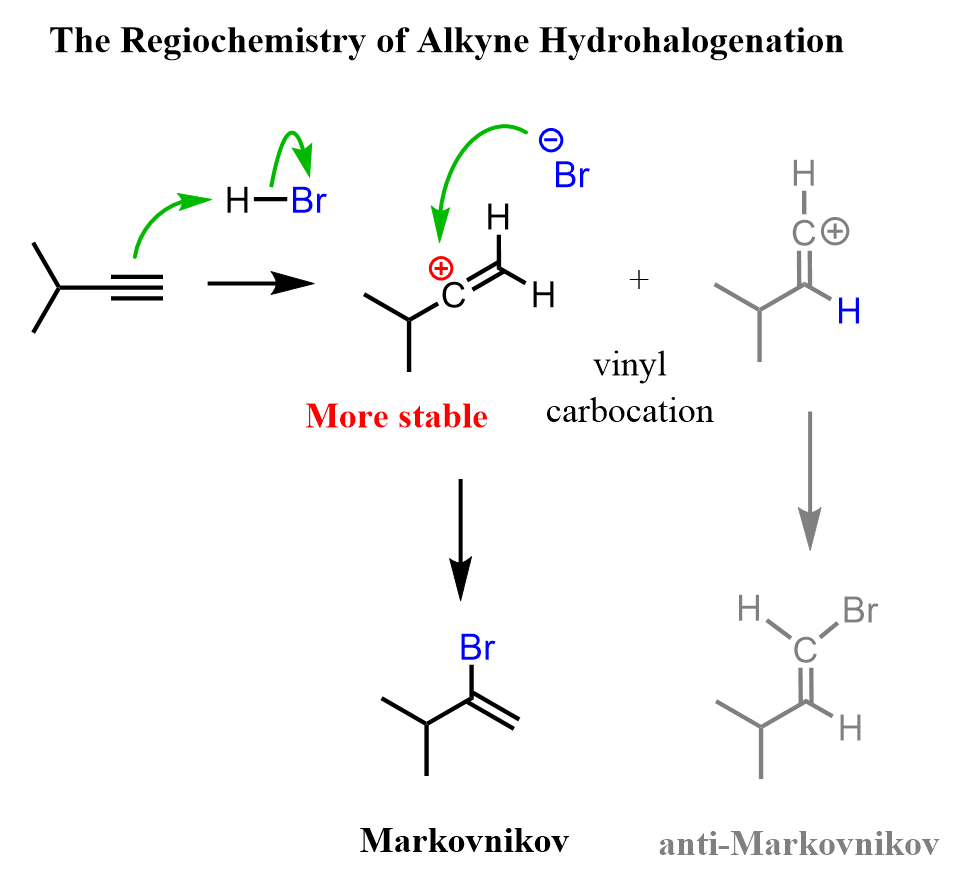
Notice that for alkynes, the regioselectivity is only relevant for terminal triple bonds where one of the carbons is connected to hydrogen. In internal alkynes, both carbons of the triple bond are connected to one carbon; we don’t have a more substituted carbon atom.
Differences in Alkenes and Alkyne Addition Reactions
As mentioned earlier, because of the extra π bond, alkynes can undergo two competitive additions when enough reactant is present. For example, when an excess of the acid is used, alkynes are converted into geminal dihalides as a result of two Markovnikov additions to the π bond:
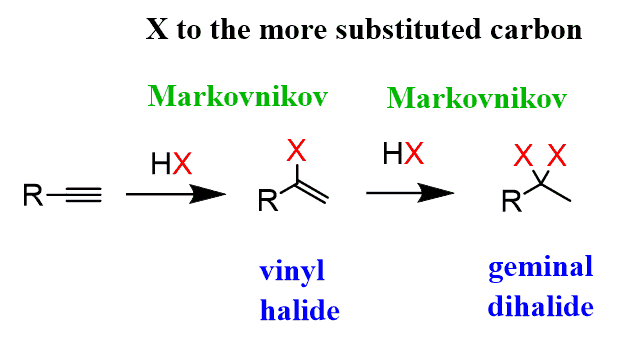
The use of excess halogen converts alkynes to tetrahalides in a similar fashion to what we saw in the halogenation of alkenes:
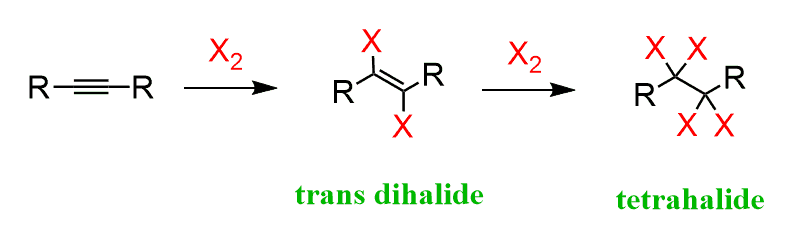
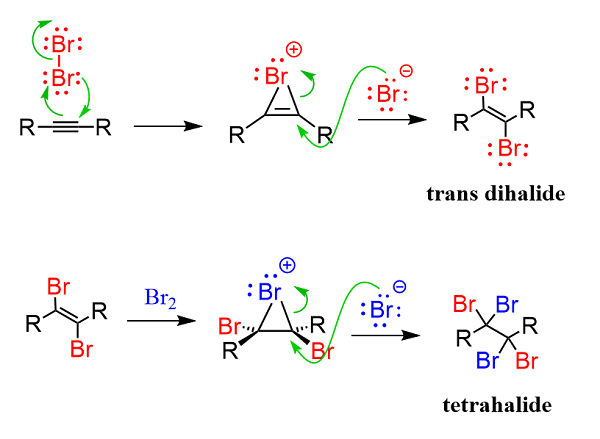
Notice that the addition of one halogen molecule forms the trans isomer of the dihalide because of the antiperiplanar addition of the halide to the intermediate halonium ion:
Hydration of Alkynes
Remember, in the article about the hydration of alkenes, we mentioned that it can be viewed as adding an H and OH groups to the carbon atoms of the double bond. As much as the general theme of the reaction is the same for alkynes, there is one significant difference: the hydration of alkynes results in an aldehyde or a ketone (not an alcohol).
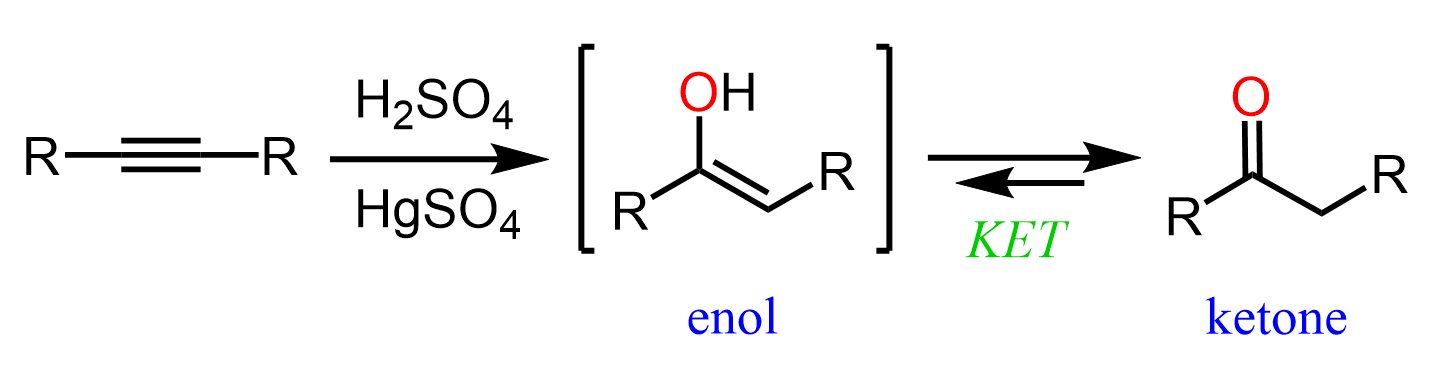
The mechanism of protonation followed by a nucleophilic attack is still the same; however, the resulting intermediate is called an enol – a molecule that combines an alkene (ene) and alcohol (ol):

Now, why is this an intermediate and the final product of the alkyne hydration reaction? We already know that alkenes are electron-rich, and it turns out that having an OH group on them increases the electron density even more via resonance donation. As a result, enols are very reactive and they convert on their own to ketones or aldehydes. This is known as keto-enol tautomerization (KET) which is a relatively complex process for this stage of the course; however, you can view it as a proton transfer from the OH group to the C=C double bond. This converts the C-OH into a C=O, and the C=C into a C-C bond:
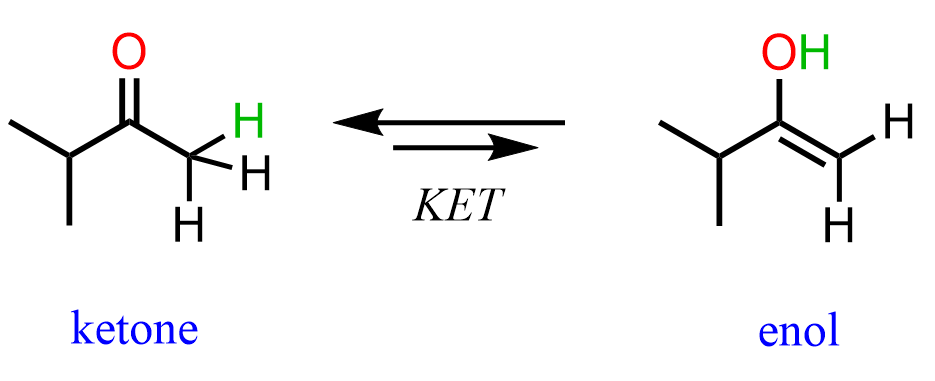
The two forms in the keto-enol tautomerization are called tautomers, and these are constitutional isomers, i.e., different compounds, so do not mix them with resonance structures.
In the next series of articles, we will discuss each of the alkyne reactions in more detail. There are also plenty of practice problems and a multiple-choice quiz on the reactions of alkynes.
Check Also
- Introduction to Alkynes
- Naming Alkynes by IUPAC Nomenclature Rules – Practice Problems
- Preparation of Alkynes by Elimination Reactions
- Hydrohalogenation of Alkynes
- Addition of Water to Alkynes
- Acid-Catalyzed Hydration of Alkynes with Practice Problems
- Reduction of Alkynes
- Halogenation of Alkynes
- Hydroboration-Oxidation of Alkynes with Practice Problems
- Ozonolysis of Alkynes with Practice Problems
- Alkylation of Terminal Alkynes in Organic Synthesis with Practice Problems
- Reactions of Acetylide Ions
- Alkyne reactions summary practice problems
- Alkyne Synthesis Reactions Practice Problems
- Alkyne Naming and Reactions Practice Quiz
- Reactions Map of Alkynes