The Stille reaction is a Pd-catalyzed cross-coupling reaction between an organostannane (organotin compound) and an organic electrophile, typically an aryl or vinyl halide (or triflate). The organostannane may contain aryl, vinyl, alkynyl, allyl, or benzyl groups, and the reaction forms a new carbon-carbon bond.
Here are some examples demonstrating how the Stille coupling is used to form new C-C bonds between sp2 and certain sp3 carbon atoms:
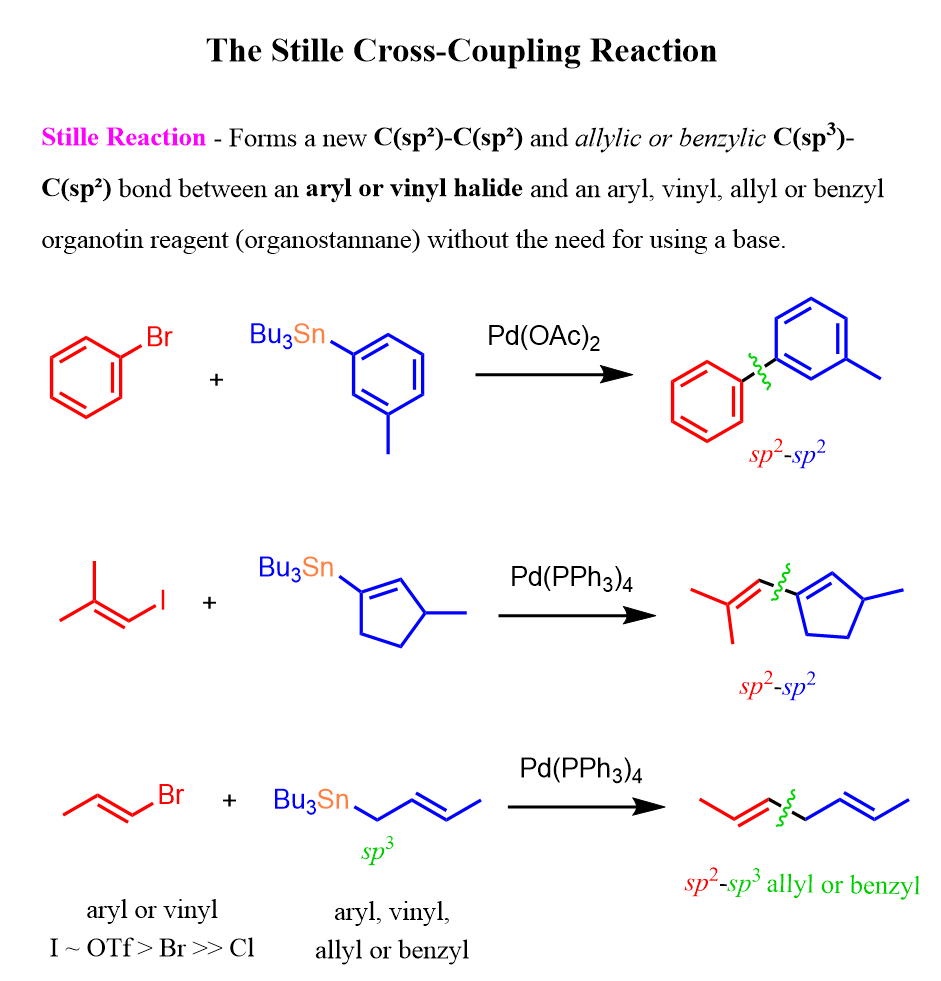
If the terms transition metal-catalyzed and cross-coupling are unfamiliar to you, be sure to check out our introductory posts on these topics, which will make understanding the Stille and other reactions of this chapter much easier.
For a short description, we can visualize the Stille coupling as a 3-major-step reaction: Oxidative addition, transmetalation, and reductive elimination.
So, let’s discuss these steps in more detail to get a complete image of the Stille coupling mechanism.
The Mechanism of the Stille Coupling Reaction
Like in other cross-coupling reactions, the first step is an oxidative addition, where the palladium catalyst inserts into the C-X bond of an organic halide (usually an aryl, vinyl, or alkyl halide), breaking the bond and forming a new Pd–C bond:
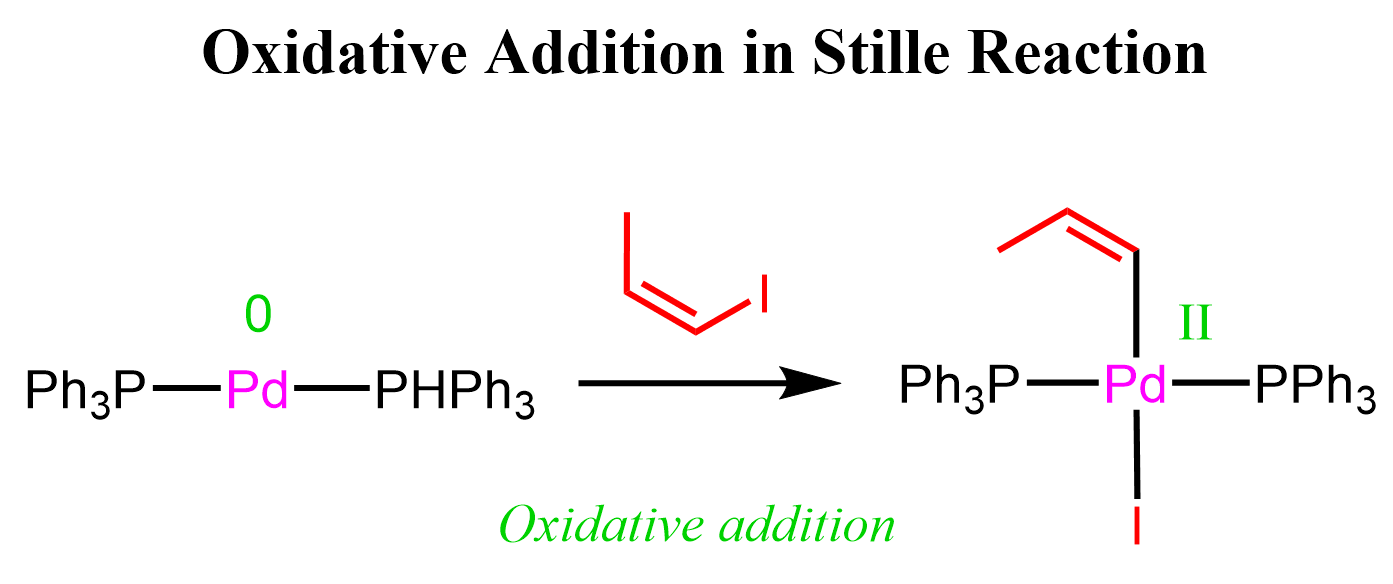
At this point, we have one of the organic fragments attached to Pd, but to couple it with the second organic partner, we need to transfer the second carbon fragment to the palladium center as well.
This happens via what is called transmetalation, which is the transfer or exchange of ligands between two metals. In the Stille coupling, the carbon fragment attached to tin is transferred from Sn to Pd in exchange for the halide ligand originally attached to the palladium:
![]()
Now, we have a new Pd intermediate that contains both carbon fragments. These two carbon groups are then coupled through the final step, called reductive elimination, which forms the new C–C bond and releases the coupled organic product:
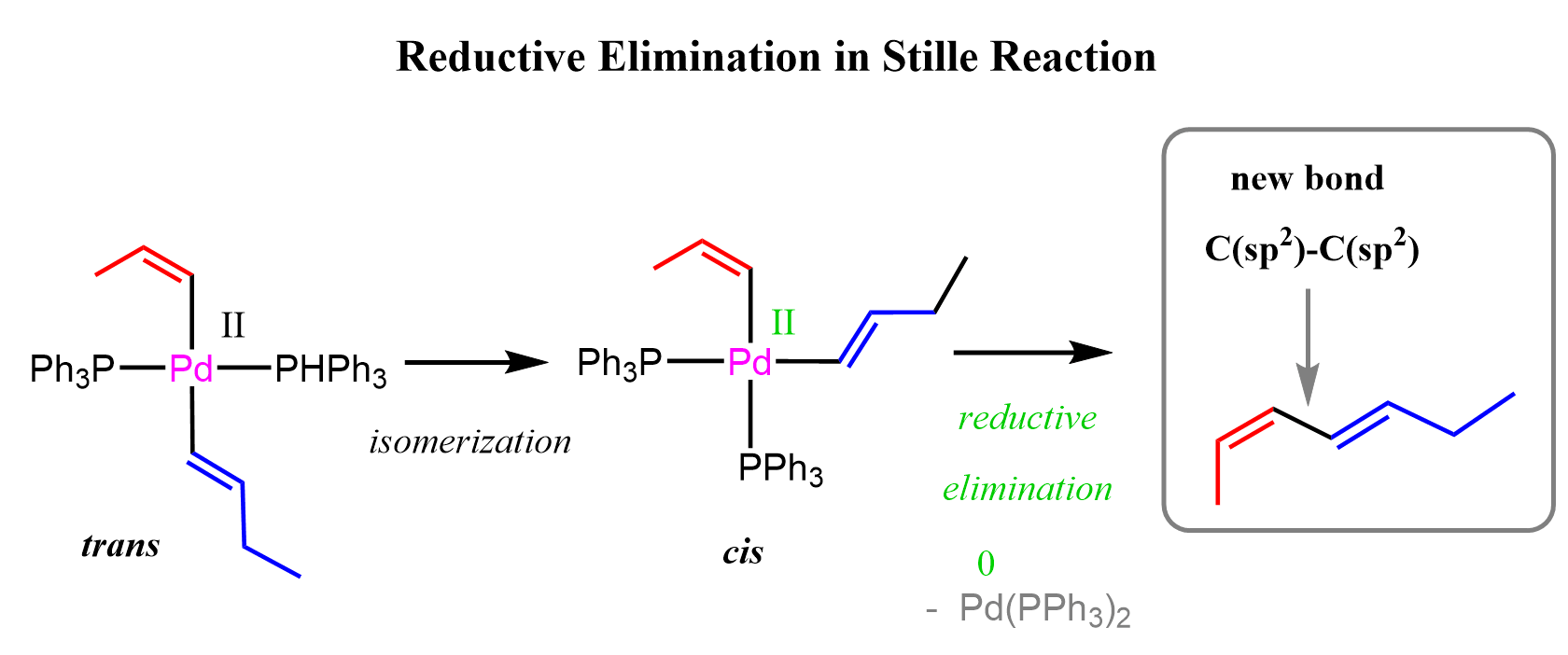
The term reductive elimination refers to the change occurring at palladium because, when the new C–C bond is formed, palladium loses both carbon ligands attached to it. As a result, the oxidation state of Pd decreases, meaning that the metal center is reduced.
One important point I wanted to mention about reductive elimination is the orientation of the two carbon groups attached to palladium. For the C–C bond formation to occur, the two carbon ligands must be positioned cis to each other.
In the cis arrangement, the two carbon groups are close enough to interact and form the new bond. If they are arranged trans to each other, the distance between them is too large, and reductive elimination is much less favorable or cannot occur directly. During the catalytic cycle, the palladium intermediate adopts the correct cis geometry via cis and trans isomerization.
The Catalytic Cycle of the Stille Coupling Reaction
So, let’s put all these steps together to show the complete mechanism of the Stille reaction in a conventional manner used for cross-coupling reactions, which is the catalytic cycle. A catalytic cycle is a simplified representation of the sequence of steps that a catalyst undergoes during a reaction. It shows how the catalyst is transformed through different intermediates, performs its function, and is eventually regenerated so that it can continue another reaction cycle:
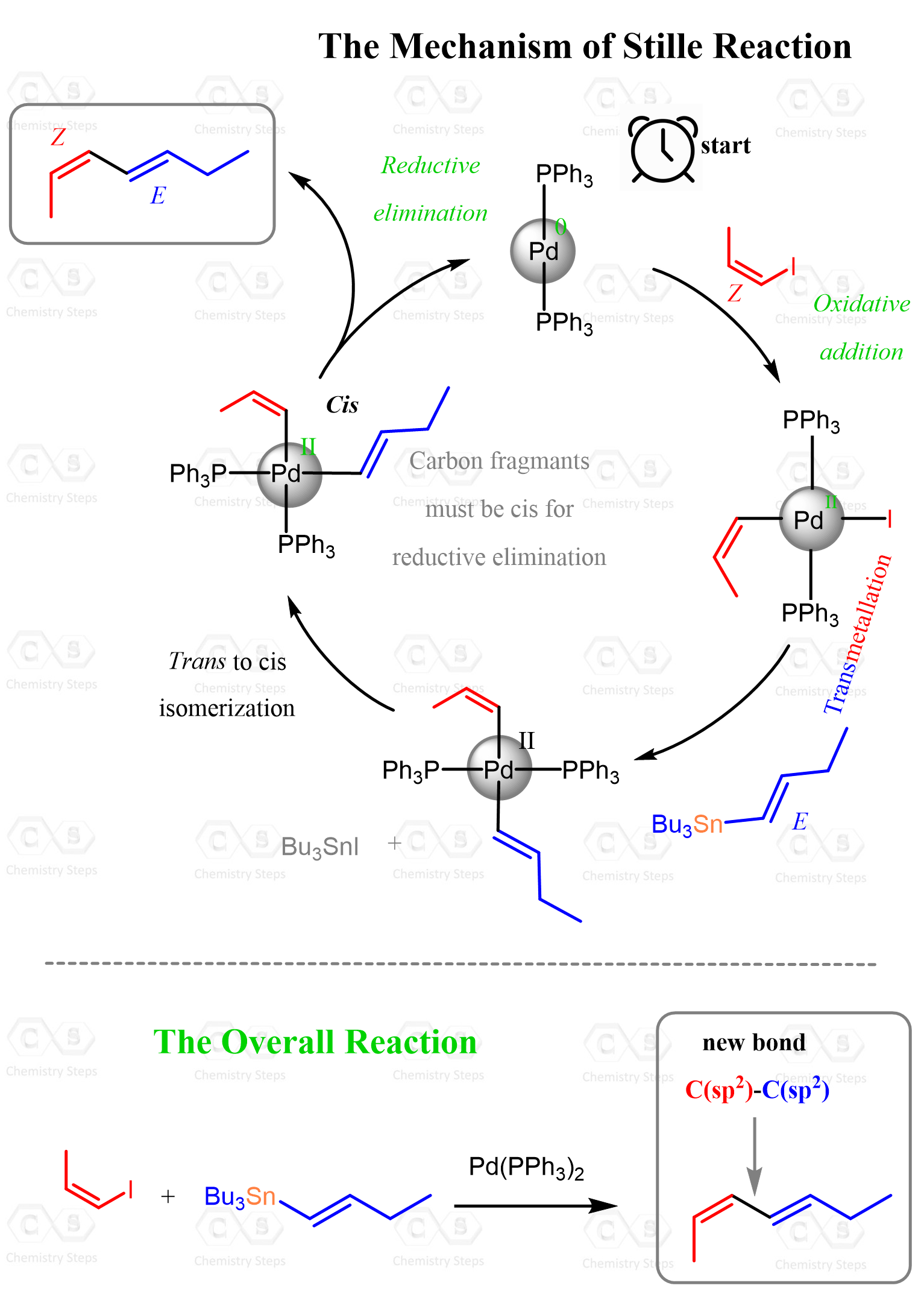
Notice how Pd(0) is converted into different palladium intermediates through oxidative addition, transmetalation, and reductive elimination, ultimately regenerating Pd(0) and allowing the process to repeat.
The Regio- and Stereoselectivity of the Stille Coupling Reaction
The Stille reaction is generally regioselective, meaning that the position of the new C–C bond is determined by the location of the halide or triflate leaving group and the C-Sn bond. In other words, the coupling occurs specifically between the carbon bearing the halide/triflate on one coupling partner and the carbon attached to tin in the organotin reagent:
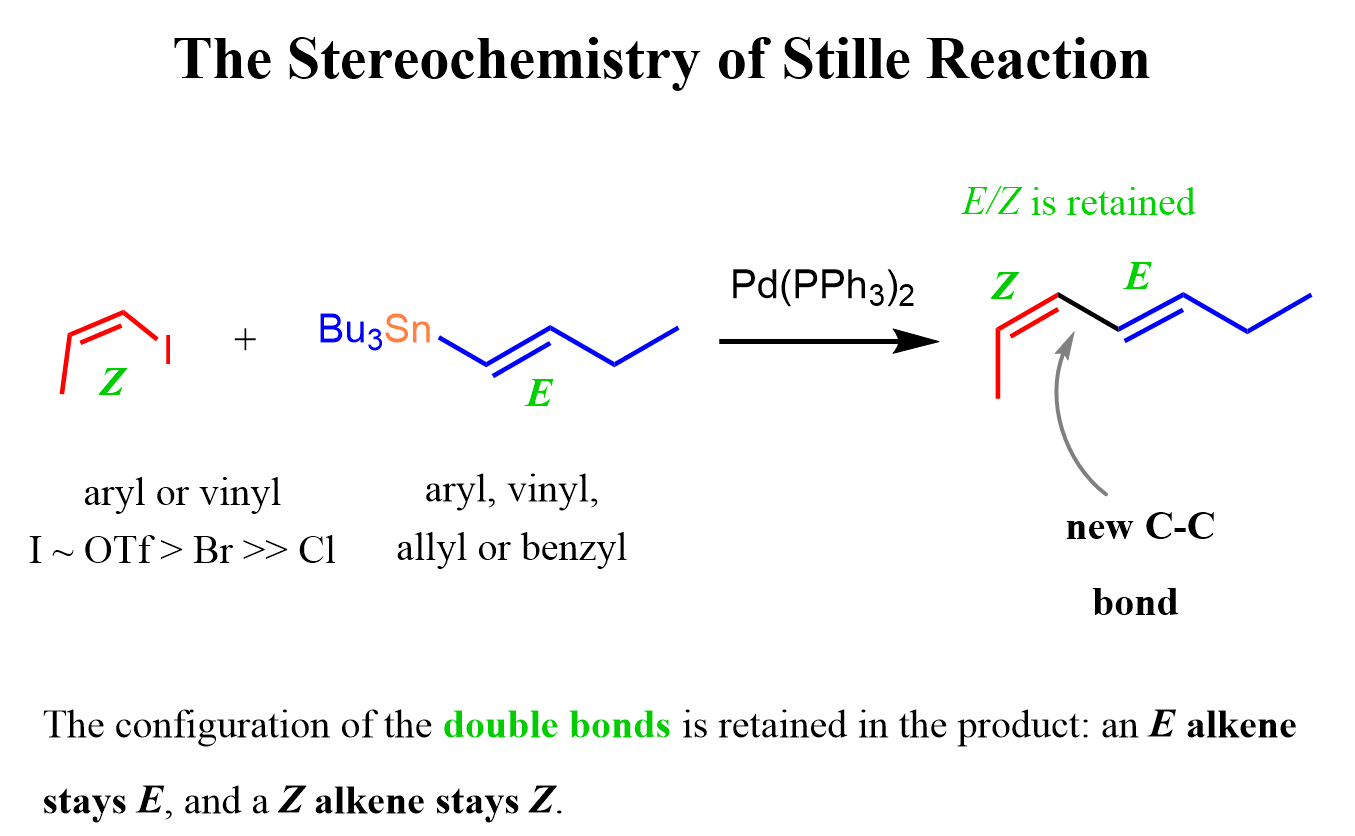
Notice also that the Stille reaction is also stereospecific, which means that the configurations of the reacting double bonds are preserved during the coupling process. Therefore, the E and Z configurations of both the organic halide and the organotin reagent are retained in the resulting coupled alkene product because the oxidative addition and transmetalation steps occur without disrupting the geometry of the C=C bonds.
The Advantages of Stille Coupling
The Stille coupling offers several important advantages that have made it a valuable cross-coupling reaction in organic synthesis.
🟢 A major advantage of the Stille reaction is the excellent functional group tolerance of organotin reagents. Organostannanes can participate in coupling reactions with aryl and vinyl halides or triflates even when other sensitive functional groups are present, including carboxylic acids, amides, esters, nitro groups, ethers, amines, hydroxyl groups, ketones, and aldehydes.
🟢 Another important advantage is the stability of organostannane reagents. Unlike many other reactive organometallic compounds, organotin reagents are relatively stable toward moisture and oxygen, allowing them to be easily prepared, isolated, and stored for extended periods.
Many organostannanes are also commercially available or can be readily prepared from Grignard reagents.
🟢 The Stille coupling also proceeds under relatively mild conditions and does not require a base, which is an advantage compared with, for example, the Suzuki coupling because of possible base-induced side reactions. In addition, not every functional group is compatible with strong basic conditions.
🟢 The Stille reaction shows high regio- and stereochemical control and can preserve the configuration of alkenyl coupling partners. This makes it a useful method for the stereoselective synthesis of complex molecules containing defined alkene geometries.
🔴 However, the Stille coupling also has some limitations. The main disadvantage is the toxicity of organotin compounds and the difficulty of removing trace amounts of tin-containing by-products from the reaction mixture. In addition, under harsh reaction conditions, some allylic and Z-alkenyl substrates can undergo double-bond migration, isomerization, and other side reactions.
🟢 To address these drawbacks of the Stille reaction, the Suzuki coupling reaction can be used as an alternative. Instead of organotin reagents, the Suzuki reaction uses organoboron compounds, such as boronic acids and boronic esters. These reagents are generally less toxic, and the boron-containing by-products are easier to remove from the reaction mixture.
📒 Overall, the Stille reaction is a palladium-catalyzed cross-coupling reaction between an organotin compound and an aryl, vinyl, or alkyl halide (or triflate), forming a new C–C bond and releasing tin-containing by-products. Its success is largely attributed to the stability of organotin reagents, mild reaction conditions, absence of a required base, and exceptional tolerance toward a wide range of functional groups.
Reference
- Milstein, D.; Stille, J. K. A General, Selective, and Facile Method for Palladium-Catalyzed Carbon–Carbon Coupling of Organotin Compounds with Organic Halides. Am. Chem. Soc. 1978, 100, 3636–3638.
- Stille, J. K. The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles. Chem. Int. Ed. Engl. 1986, 25, 508–524.
- Mitchell, T. N. Palladium-Catalysed Reactions of Organotin Compounds. Synthesis 1992, 803–815.
- Farina, V.; Krishnamurthy, V.; Scott, W. J. The Stille Reaction. React. 1997, 50, 1–652.
- Kosugi, M.; Fugami, K. Review of Stille-Type Coupling Reactions. Organomet. Chem. 2002, 653, 50–53.
- Espinet, P.; Echavarren, A. M. The Mechanisms of the Stille Reaction. Chem. Int. Ed. 2004, 43, 4704–4734.
- Cordovilla, C.; Bartolomé, C.; Martínez-Ilarduya, J. M.; Espinet, P. The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053.
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, 2012.
- László Kürti and Barbara Czakó, Strategic Applications of Named Reactions in Organic Synthesis, 2005